
前言
中(zhōng)美藥品監管機構通過設定加快上市程序(Expedited Programs)為(wèi)那些具(jù)有(yǒu)滿足臨床急需醫(yī)療需求潛力或是有(yǒu)充分(fēn)證據表明優于臨床現有(yǒu)療法的藥品提供了上市加速渠道,以及相應的技(jì )術和政策支持。數據表明,上市加速程序的加速效果顯著,其中(zhōng),NMPA的優先審評審批政策可(kě)将藥物(wù)審評時間從406天縮減至77天;FDA的突破性療法認定則可(kě)将藥物(wù)的開發時間從8年縮短至4.8年。
為(wèi)了加快藥物(wù)開發和上市,加速滿足臨床急需醫(yī)療需求,包括NMPA、FDA、EMA等在内的各國(guó)藥品監管機構均頒布了相關加快上市程序。其中(zhōng),NMPA在2020版的《藥品注冊管理(lǐ)辦(bàn)法》中(zhōng),設立了“突破性治療藥物(wù)、附條件批準、優先審評審批、特别審批”四個加快上市程序,FDA則通過立法設立了“快速通道(Fast Track)、突破性療法(Breakthrough Therapy)、再生醫(yī)學(xué)先進療法(Regenerative Medicine Advanced Therapy)、優先審評(Priority Review)、加速審批(Accelerated Approval)”五個加快上市程序。NMPA在2021年共批準了75個新(xīn)藥,其中(zhōng)16個新(xīn)藥通過優先審評審批和附條件批準加速上市,在獲批的15款國(guó)産(chǎn)創新(xīn)藥中(zhōng),更是有(yǒu)高達14款獲得了優先審評審批或附條件批準,同年FDA批準的新(xīn)藥中(zhōng)則是有(yǒu)71%通過優先審評批準上市,足以見得申請加快上市程序對創新(xīn)藥開發的重要性。
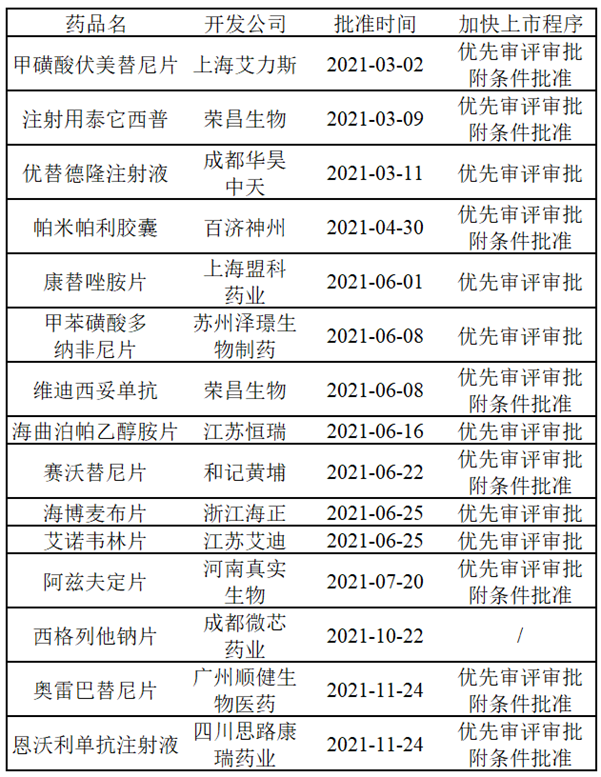
那麽什麽樣的新(xīn)藥才能(néng)申請到加快上市程序?申請到加快上市程序又(yòu)能(néng)多(duō)大程度地加速新(xīn)藥上市呢(ne)?根據NMPA頒布的《藥品注冊管理(lǐ)辦(bàn)法》,将加快上市程序的相關特性總結如下:
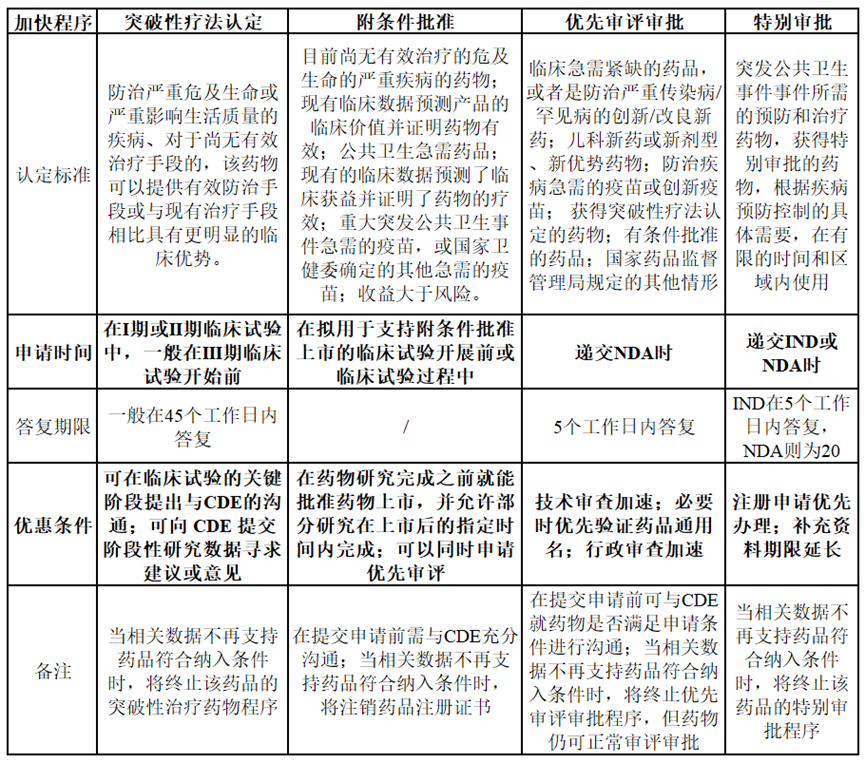
可(kě)以發現,能(néng)夠獲得加快上市的藥物(wù)均是具(jù)有(yǒu)滿足臨床急需醫(yī)療需求的潛力或是有(yǒu)充分(fēn)證據表明優于臨床現有(yǒu)療法,藥品獲得優先審評審批或突破性療法認定後,可(kě)獲得來自NMPA的政策和技(jì )術支持,包括但不限于必要的技(jì )術指導、與藥品申請人的溝通、資源的優先分(fēn)配和縮短審查時間等。
由百濟神州開發的澤布替尼(zanubrutinib)便是納入加快上市程序的典型。一項入組86例患者的單臂II期臨床試驗結果顯示,在複發性和/或難治性套細胞淋巴瘤患者中(zhōng),澤布替尼的ORR率為(wèi)83.7%,CR達到78%,房顫、第二腫瘤、腫瘤溶解綜合征的發生率均為(wèi)0,
療效和安(ān)全性均優于既往獲批的BTK抑制劑,基于此,澤布替尼先後獲得了NMPA的突破性療法認定、優先審評審批以及附條件批準。更早之前同樣也獲得了FDA的突破性療法認定以及加速批準,從遞交NDA申請到加速批準上市僅耗時5個月。
特别審批程序則主要适用(yòng)于突發公(gōng)共衛生事件所需藥物(wù),如COVID-19,2021年NMPA審結了81件納入特别審批程序的注冊申請(包括IND和NDA),均為(wèi)新(xīn)冠病毒疫苗和治療藥物(wù),并建議附條件批準5件新(xīn)冠病毒疫苗NDA。
根據NMPA的2021年度藥品審評報告,我們也能(néng)一探各個加快上市程序申請的成功率。NMPA在2021年共受理(lǐ)突破性治療藥物(wù)程序的注冊申請263件,批準了其中(zhōng)的53件,成功率為(wèi)20.1%;受理(lǐ)NDA共389件,其中(zhōng)有(yǒu)115件納入了優先審評審批程序,占比29.5%。可(kě)以看出雖然成功率并不算高,但相比去年仍有(yǒu)明顯提高,一方面說明中(zhōng)國(guó)新(xīn)藥的創新(xīn)水平正不斷提升,另一方面也說明藥審中(zhōng)心的審評資源在逐年向具(jù)有(yǒu)臨床優勢的新(xīn)藥注冊申請傾斜。
FDA的加速政策與NMPA的相關政策大同小(xiǎo)異,此處不再贅述。對于藥品申請人來說,最關心的可(kě)能(néng)還是獲得監管機構的加快上市認定後,到底能(néng)節約多(duō)長(cháng)時間,可(kě)為(wèi)自己的産(chǎn)品帶來多(duō)大的先發優勢。
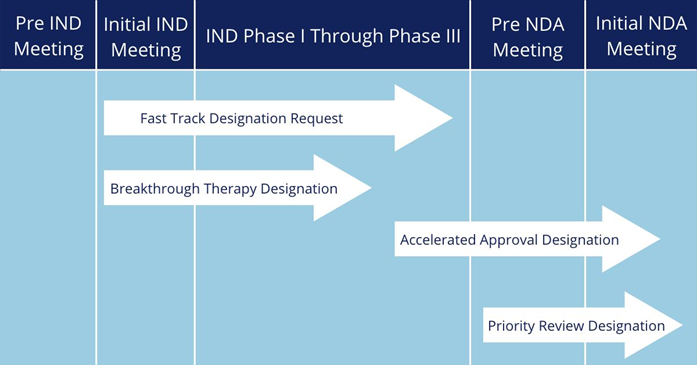
FDA加快上市程序
根據NMPA的規定,納入優先審評審批程序的藥品上市許可(kě)申請,審評時限為(wèi)130日,其中(zhōng)臨床急需的境外已上市境内未上市的罕見病藥品審評時限為(wèi)70日,而普通NDA的審評時限則為(wèi)200天。
另外,根據中(zhōng)國(guó)醫(yī)學(xué)科(kē)學(xué)院腫瘤醫(yī)院的研究報告,從2016年到2020年,獲得優先審評審批資格的藥品其中(zhōng)位審評時間僅為(wèi)77天,而對普通審評審批的藥品說,遞交NDA後耗時的中(zhōng)位數為(wèi)406天,顯著長(cháng)于納入優先審評審批程序的藥品。
其他(tā)各國(guó)的藥品監管機構的加快上市程序有(yǒu)着類似的效果。對于未獲得優先審評資格的藥品來說,
在2021年,從向FDA遞交NDA到獲得批準上市的中(zhōng)位時長(cháng)為(wèi)365天,而優先審評的藥品則可(kě)将這一時間縮短123天,大大加速創新(xīn)藥物(wù)的上市及臨床應用(yòng)。其他(tā)國(guó)家/地區(qū)如歐盟、日本、加拿(ná)大等的優先審評政策也均可(kě)縮短藥品審評審批耗時26%-46%不等。
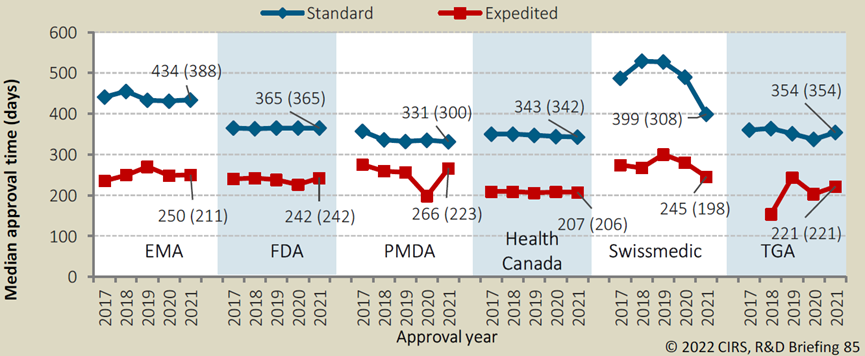
各國(guó)監管優先審評vs普通審評耗時
事實上,獲得加快上市相關政策資格後,藥物(wù)的臨床開發時間同樣可(kě)以得到節約。根據發表在JAMA的相關文(wén)章,獲得一項FDA加快上市政策資格的藥品中(zhōng)位開發時間(從IND申請到FDA首次批準的時間)為(wèi)7.1年,相比沒有(yǒu)任何加快上市政策資格的藥品縮短了0.9年;盡管優先審評資格能(néng)夠将藥品的審評時間縮短4個月,但令人詫異的是,對于非突破性、非快速通道的藥物(wù),即使納入優先審評或加速批準,他(tā)們與未獲得加快上市資格的藥物(wù)在開發時間上并沒有(yǒu)顯著差異;而對于被授予突破性療法認定或快速通道資格的藥品來說,開發時間則明顯縮短,他(tā)們的中(zhōng)位開發時間分(fēn)别隻有(yǒu)4.8年和7.0年,尤其是突破性療法認定,将藥品的開發時間整整減少了40%,充分(fēn)體(tǐ)現了突破性療法認定的優勢,更及時地為(wèi)患者送去了急需新(xīn)藥,也為(wèi)藥企赢得了更長(cháng)的上市獨占期。
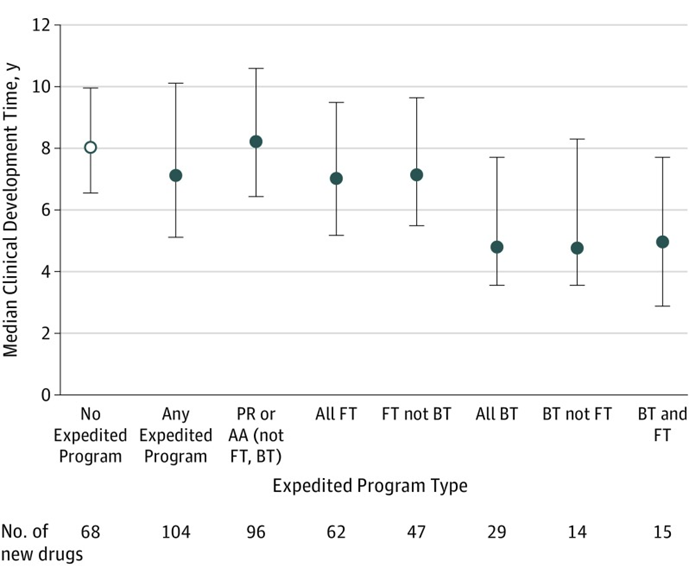
FDA不同加快上市政策的中(zhōng)位開發時間
參考:
[1] 《Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics》
[2] 《藥品注冊管理(lǐ)辦(bàn)法》
[3] Wang S, Yang Q, Deng L. An overview of cancer drugs approved through expedited approval programs and orphan medicine designation globally between 2011 and 2020. Drug Discov Today. 2022 May;27(5):1236-1250.
[4] Hwang TJ, Darrow JJ, Kesselheim AS. The FDA's Expedited Programs and Clinical Development Times for Novel Therapeutics, 2012-2016. JAMA. 2017 Dec 5;318(21):2137-2138.
[5] 《2021年度藥品審評報告》
[6] https://www.fda.gov/
----------THE END----------
免責聲明:本文(wén)系轉載分(fēn)享,文(wén)章觀點、内容、圖片及版權歸原作(zuò)者所有(yǒu),如涉及侵權請聯系删除!