撰文(wén):克裏斯
IF:50
推薦度:⭐⭐⭐⭐⭐
亮點:
文(wén)章揭示了胰腺癌細胞和巨噬細胞之間的新(xīn)型正反饋循環,該循環通過促進TWEAK分(fēn)泌加劇了胰腺癌的惡病質(zhì)。研究發現巨噬細胞的耗竭和TWEAK的抑制可(kě)能(néng)是治療胰腺癌惡病質(zhì)的有(yǒu)效靶點。此外,研究還強調了CCL2和CCL5在這一過程中(zhōng)的關鍵作(zuò)用(yòng),為(wèi)未來的治療策略提供了分(fēn)子層面的見解。研究發現不僅具(jù)有(yǒu)臨床相關性,還展示了向臨床轉化的潛力,為(wèi)胰腺癌惡病質(zhì)的治療提供了新(xīn)的方向。
2024年4月11日,美國(guó)俄克拉荷馬大學(xué)健康科(kē)學(xué)中(zhōng)心醫(yī)學(xué)系Mingyang Liu研究團隊在國(guó)際知名(míng)期刊《Cancer cell》發表了題為(wèi):The crosstalk between macrophages and cancer cells potentiates pancreatic cancer cachexia的研究文(wén)章。

胰腺癌相關的癌性惡病質(zhì)(pancreatic cancer cachexia),這是一種嚴重的消耗性疾病,與體(tǐ)重下降、食欲減退和肌肉消耗有(yǒu)關。胰腺癌是癌症死亡的常見原因,且預後極差,因為(wèi)大多(duō)數患者在診斷時已處于晚期,此時治療選擇有(yǒu)限。癌性惡病質(zhì)對患者的生活質(zhì)量和總體(tǐ)生存率有(yǒu)着顯著的負面影響,但目前尚無批準的治療方案。
文(wén)章強調了研究腫瘤細胞與免疫微環境之間相互作(zuò)用(yòng)的重要性,特别是巨噬細胞在胰腺癌進展和惡病質(zhì)中(zhōng)的作(zuò)用(yòng)。巨噬細胞是胰腺癌免疫微環境中(zhōng)最豐富的免疫細胞類型之一,與癌症的進展和轉移有(yǒu)關。研究者們指出,盡管炎症是癌症惡病質(zhì)的關鍵特征之一,但腫瘤微環境如何調控肌肉消耗的機制尚不清楚。因此,本研究旨在探索巨噬細胞在胰腺癌引起的肌肉消耗中(zhōng)的作(zuò)用(yòng),以及它們如何通過分(fēn)泌特定因子(如TWEAK)來促進這一過程。通過理(lǐ)解這些相互作(zuò)用(yòng),研究者們希望識别出潛在的治療靶點,以開發更有(yǒu)效的治療胰腺癌惡病質(zhì)的方法。
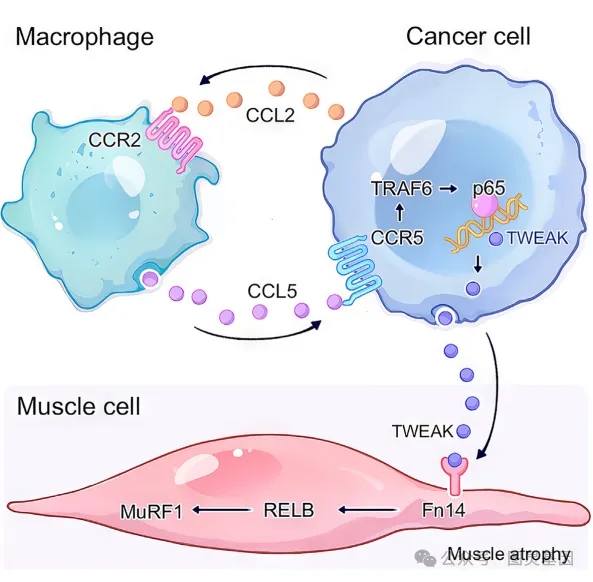
圖1、癌細胞、巨噬細胞、肌肉細胞之間的相互作(zuò)用(yòng)
1、巨噬細胞在促進肌肉消耗和癌症惡病質(zhì)中(zhōng)的作(zuò)用(yòng)
巨噬細胞在胰腺癌惡病質(zhì)中(zhōng)能(néng)夠促進肌肉消耗,研究員通過分(fēn)析不同癌症類型的數據,發現巨噬細胞的浸潤與惡病質(zhì)的發生率和體(tǐ)重下降正相關。在胰腺癌小(xiǎo)鼠模型中(zhōng),巨噬細胞的耗竭能(néng)夠減輕肌肉萎縮,提高肌肉力量,并減少體(tǐ)重損失。此外,TWEAK(一種凋亡誘導因子)的表達在巨噬細胞的作(zuò)用(yòng)下上調,并通過激活肌肉重塑的MuRF1途徑促進肌肉萎縮。胰腺癌細胞與巨噬細胞的相互作(zuò)用(yòng)導緻TWEAK的非自主分(fēn)泌增加,這一過程涉及CCL5/TRAF6/NF-kB信号通路。研究還發現,通過CCL2/CCR2軸,腫瘤細胞能(néng)夠招募并重編程巨噬細胞,這一過程對肌肉萎縮的發展至關重要。這些發現表明,巨噬細胞和TWEAK是治療胰腺癌惡病質(zhì)的潛在治療靶點。
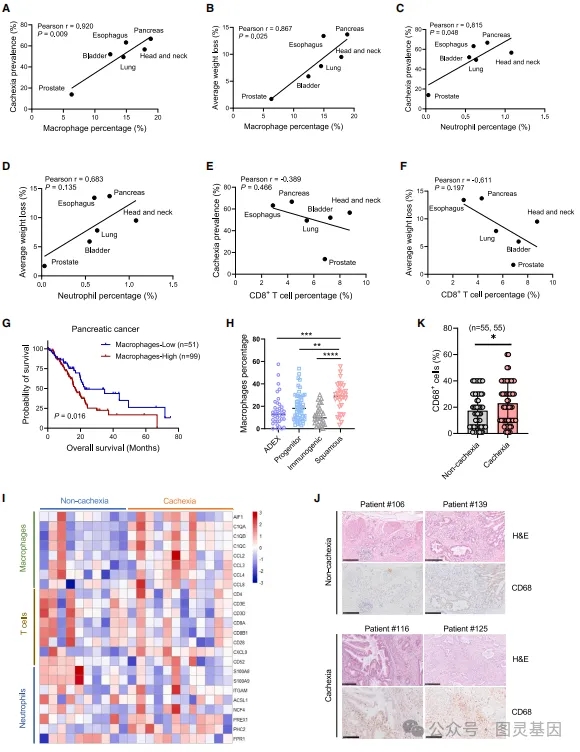
圖2、腫瘤相關巨噬細胞促進肌肉萎縮和腫瘤惡病質(zhì)
2、耗竭巨噬細胞減輕胰腺癌小(xiǎo)鼠肌肉萎縮
研究顯示,在胰腺癌小(xiǎo)鼠模型中(zhōng),巨噬細胞的耗竭能(néng)夠顯著減輕肌肉萎縮。通過遺傳敲除(Ccr2-/-小(xiǎo)鼠模型)或藥物(wù)處理(lǐ)法耗竭巨噬細胞,小(xiǎo)鼠的肌肉力量得到增強,體(tǐ)重下降減少,肌肉重量增加,且肌肉萎縮标記物(wù)MuRF1和Atrogin-1的表達降低。結果證實了巨噬細胞在胰腺癌引起的肌肉萎縮中(zhōng)起重要作(zuò)用(yòng),并提示巨噬細胞耗竭策略可(kě)能(néng)是治療胰腺癌惡病質(zhì)的潛在方法。
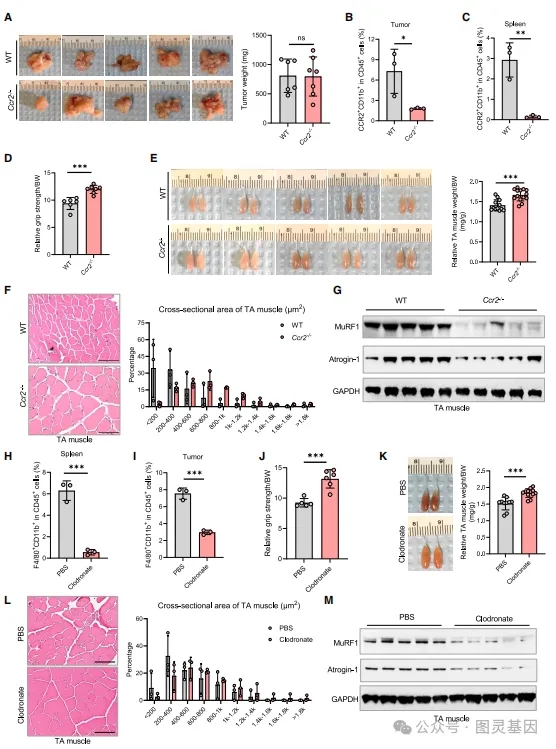
圖3、巨噬細胞減少減輕胰腺癌小(xiǎo)鼠模型肌肉萎縮
3、巨噬細胞上調TWEAK表達促進胰腺癌中(zhōng)肌肉消耗機制
在胰腺癌中(zhōng),巨噬細胞通過促進腫瘤壞死因子超家族成員12(TWEAK)的表達來加劇肌肉消耗。研究發現,TWEAK的表達與M2型巨噬細胞的浸潤密切相關,且TWEAK能(néng)夠激活C2C12分(fēn)化肌管中(zhōng)的肌肉萎縮标記物(wù)Atrogin-1和MuRF1,以及RELB信号通路。巨噬細胞與胰腺癌細胞共培養時,TWEAK的分(fēn)泌顯著增加,表明巨噬細胞能(néng)以非自主方式促進TWEAK的分(fēn)泌。此外,巨噬細胞分(fēn)泌的CCL5通過激活NF-kB信号通路,特别是p65亞單位,進一步促進TWEAK的表達。ChIP實驗和熒光素酶報告基因分(fēn)析證實了p65可(kě)以直接激活TWEAK的轉錄。通過敲低CCR5或使用(yòng)NF-kB信号通路的小(xiǎo)分(fēn)子抑制劑,可(kě)以抑制巨噬細胞誘導的TWEAK表達增加,揭示了CCL5/CCR5軸和NF-kB信号通路在TWEAK表達調控中(zhōng)的作(zuò)用(yòng)。
4、胰腺癌細胞招募巨噬細胞誘導TWEAK激活TRAF6/p65通路
胰腺癌細胞通過CCL2信号軸招募巨噬細胞,這些巨噬細胞在腫瘤微環境中(zhōng)促進TWEAK(腫瘤壞死因子超家族成員12)的分(fēn)泌。TWEAK是一種能(néng)夠引起肌肉萎縮的細胞因子,其在胰腺癌中(zhōng)的表達通過CCL5/TRAF6/NF-kB信号通路被巨噬細胞激活。巨噬細胞分(fēn)泌的CCL5促進了胰腺癌細胞中(zhōng)TWEAK的表達,這一過程涉及TRAF6介導的K63泛素化和p65的激活。TWEAK的增加進一步激活了肌肉細胞中(zhōng)的MuRF1信号通路,導緻肌肉萎縮和惡病質(zhì)。
5、胰腺癌中(zhōng)CCL2激活巨噬細胞誘導肌肉萎縮
CCL2由胰腺癌細胞産(chǎn)生,招募并激活巨噬細胞,這些巨噬細胞通過增加肌肉萎縮标志(zhì)物(wù)MuRF1和Atrogin-1的表達,促進了肌肉萎縮。此外,CCL2激活的巨噬細胞還增加了TWEAK的表達,TWEAK是一種關鍵的肌肉萎縮因子。這一過程涉及NF-kB信号通路的激活,以及ZXDC轉錄因子對CCL2表達的調控。
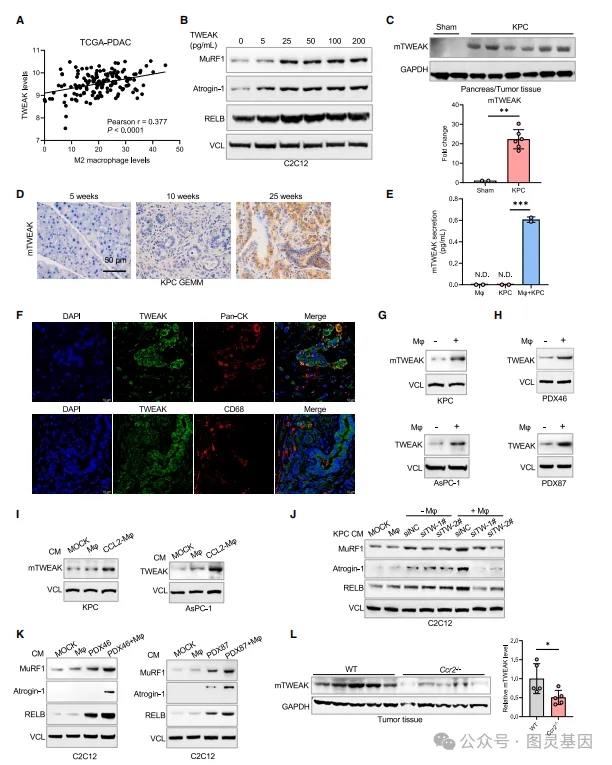
圖4、胰腺癌中(zhōng)巨噬細胞通過上調TWEAK促進肌肉萎縮
6、癌症誘導的肌肉萎縮部分(fēn)依賴于TWEAK的非自主激活
胰腺癌通過促進腫瘤細胞與巨噬細胞之間的相互作(zuò)用(yòng),引起TWEAK(腫瘤壞死因子超家族成員12)的非自主激活,這一過程在體(tǐ)外和體(tǐ)内模型中(zhōng)均被證實可(kě)增加肌肉萎縮标志(zhì)物(wù)MuRF1和Atrogin-1的表達。在臨床樣本中(zhōng),惡病質(zhì)患者的腫瘤組織顯示TWEAK和巨噬細胞标記物(wù)CD68的表達水平更高,肌肉組織中(zhōng)MuRF1的表達也随之增加,揭示了TWEAK在胰腺癌惡病質(zhì)中(zhōng)的重要作(zuò)用(yòng)。這些發現表明,TWEAK的非自主激活部分(fēn)依賴于腫瘤微環境中(zhōng)的巨噬細胞,為(wèi)開發針對胰腺癌惡病質(zhì)的治療策略提供了新(xīn)的靶點。
教授介紹

Mingyang Liu,現擔任俄克拉荷馬大學(xué)健康科(kē)學(xué)中(zhōng)心(University of Oklahoma Health Sciences Center)研究員,在該中(zhōng)心的外科(kē)部門工(gōng)作(zuò)。劉博士的研究興趣包括胰腺癌、手性、超分(fēn)子化學(xué)以及腫瘤微環境,特别關注CCL5這一領域。在學(xué)術工(gōng)作(zuò)和影響力方面,劉博士發表了49篇論文(wén),其合作(zuò)跨越了多(duō)個機構和部門,展示了在研究上的跨學(xué)科(kē)方法。與多(duō)位研究人員合作(zuò)過,在業内獲得廣泛認可(kě),論文(wén)獲得的引用(yòng)次數超1300次。
參考文(wén)獻
Liu M, Ren Y, Zhou Z, Yang J, Shi X, Cai Y, Arreola AX, Luo W, Fung KM, Xu C, Nipp RD, Bronze MS, Zheng L, Li YP, Houchen CW, Zhang Y, Li M. The crosstalk between macrophages and cancer cells potentiates pancreatic cancer cachexia. Cancer Cell. 2024 Mar 29:S1535-6108(24)00094-1. doi: 10.1016/j.ccell.2024.03.009. Epub ahead of print. PMID: 38608702.
----------THE END----------免責聲明:本文(wén)系轉載分(fēn)享,文(wén)章觀點、内容、圖片及版權歸原作(zuò)者所有(yǒu),如涉及侵權請聯系删除!
Copyright © 2017 京ICP證000000号